Targeting RNase H2 Exposes a New Therapeutic Vulnerability in Triple-Negative Breast Cancer
6 May 2026
Key Takeaways
- Researchers at MD Anderson have identified RNase H2 as a key survival mechanism in triple-negative breast cancer, helping tumor cells tolerate high levels of DNA replication stress.
- In preclinical models, inhibiting RNase H2 increased DNA damage, suppressed tumor growth, and activated innate immune signals that recruited T cells.
- The strategy also enhanced responses to ATR, PARP, and immune checkpoint inhibitors, providing a rationale for future combination strategies.
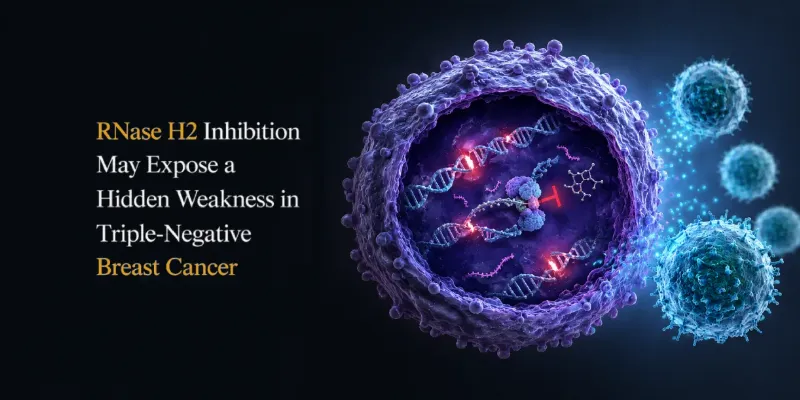
Triple-negative breast cancer is often described by what it lacks: estrogen receptor, progesterone receptor, and HER2. But a new preclinical study from The University of Texas MD Anderson Cancer Center, published in Cell Reports Medicine, suggests that this aggressive subtype may also be understood by what it depends on to survive: an enzyme called RNase H2. By blocking RNase H2, researchers increased DNA damage inside TNBC cells and activated immune signals that may help recruit T cells into the tumor microenvironment.
“Adding RNase H2 inhibition is a one-two punch that overcomes the adaptive mechanism that triple-negative breast cancer tumors leverage to survive replication stress and continue progressing,” said Shiaw-Yih Lin, Ph.D., professor of Systems Biology at MD Anderson. “Our findings show that this is a promising therapeutic strategy that lays the groundwork to meaningfully improve patient outcomes for this aggressive and difficult-to-treat subtype of breast cancer.”
Turning a survival mechanism against the tumor
Many cancer treatments, including several used in breast cancer, work by pushing tumor cells into DNA damage. In theory, cells under excessive replication stress should die. Yet TNBC cells often survive despite high genomic instability, rapid proliferation, and defective DNA repair pathways.
The MD Anderson team, led by professor Shiaw-Yih Lin, focused on one possible explanation: TNBC cells may adapt to replication stress by increasing their reliance on RNase H2. This enzyme normally removes ribonucleotides, RNA-like building blocks, that are mistakenly inserted into genomic DNA during replication. If these embedded RNA fragments are not removed, they can destabilize DNA and contribute to cell death.
The researchers found that RNASEH2A, the catalytic subunit of RNase H2, is overexpressed in TNBC tumors and is associated with poor survival. In experimental models, RNASEH2A overexpression appeared to allow cells to escape oncogene-induced senescence and continue proliferating under conditions of replication stress.
When the investigators genetically silenced RNASEH2A, or pharmacologically inhibited RNase H2, TNBC cells accumulated more replication stress and DNA damage. Importantly, this effect selectively impaired TNBC viability while sparing non-tumorigenic mammary epithelial cells in preclinical models. Tumor growth was also suppressed in vivo.
A DNA damage strategy with an immune effect
The study’s most important message is not simply that RNase H2 inhibition damages TNBC cells. It is that the same process may also help make tumors more visible to the immune system.
Blocking RNase H2 led to accumulation of cytosolic single-stranded DNA, a danger signal that can activate innate immune pathways. This activation increased the expression of chemokines involved in recruiting T cells, potentially reshaping the tumor microenvironment in favor of immune attack.
That dual mechanism matters because TNBC has been one of the more immunologically active breast cancer subtypes, yet immune checkpoint inhibitors benefit only a subset of patients. Strategies that increase immune visibility while simultaneously weakening tumor-cell survival could be especially valuable.
In the study, RNase H2 inhibition enhanced the efficacy of immune checkpoint blockade in preclinical models. This suggests a potential path toward combination strategies that link DNA damage modulation with immunotherapy.
Why combinations may be central
The findings also point to possible synergy with two major classes of DNA damage response therapies: ATR inhibitors and PARP inhibitors.
ATR helps cells respond to replication stress, while PARP inhibitors exploit defects in DNA repair. By increasing replication stress and DNA damage, RNase H2 inhibition may make TNBC cells more vulnerable to both approaches. In the study, RNase H2 inhibition sensitized TNBC models to ATR and PARP inhibitors.
This is clinically relevant because TNBC is biologically heterogeneous. Some tumors may be more dependent on replication stress response pathways, others may be more sensitive to immune modulation, and some may require rational combinations. RNase H2 inhibition could, in principle, be paired with DNA damage response agents, immune checkpoint blockade, or both, depending on tumor biology and patient-specific factors.
Still early, but biologically compelling
Despite its highly interesting findings, this work remains preclinical, and no conclusions can yet be drawn about safety, efficacy, dose, biomarkers, or patient selection in humans. Even so, the biological rationale is strong. TNBC cells appear to survive by managing the consequences of their own genomic instability. RNase H2 helps remove RNA fragments embedded in DNA, reducing lethal stress. Inhibiting the enzyme removes that protection, intensifies DNA damage, and may trigger immune pathways that attract T cells.
For a cancer subtype still lacking enough effective targeted options, that combination is notable. Rather than attacking TNBC through a single pathway, RNase H2 inhibition may exploit a core survival adaptation while simultaneously opening the door to stronger antitumor immunity. If confirmed in clinical studies, this approach could add a new layer to TNBC treatment: not only damaging the tumor, but also helping the immune system recognize the damage.





Comments
No Comments Yet!